1.本发明涉及一种分子筛及其制备方法和应用。
背景技术:
2.在工业上,分子筛材料因其具有空旷的结构和大的表面积而被广泛用于催化、离子交换、吸附和分离等领域。这些材料结构上的细微差别,预示着用来表征它们的各种可观察性能有差异,如它们的形貌、比表面积、空隙尺寸和这些尺寸的可变性等,同时也意味着它们本身在材料的催化和吸附性能等方面存在重大差异。
3.结晶微孔沸石的基本骨架结构是基于刚性的三维to4(sio4,alo4等)单元结构;在此结构中to4是以四面体方式共享氧原子,骨架四面体如alo4的电荷平衡是通过表面阳离子如na
+
、h
+
的存在保持的。由此可见通过阳离子交换方式可以改变沸石的骨架性质。同时,在沸石的结构中存在着丰富的、孔径一定的孔道体系,这些孔道相互交错形成三维网状结构,且孔道中的水或有机物被去除后其骨架仍能稳定存在(us 4439409)。正是基于上述结构,沸石不但对多种有机反应具有良好催化活性、优良的择形性、并通过改性可实现良好的选择性(us 6162416,us 4954325,us 5362697)。
4.具有mww结构分子筛具有两套独立的互不相通的十元环孔道体系:一套二维正弦孔道,孔道截面近似椭圆形,孔径为0.41nm
×
0.51nm,另一套十元环孔道含有尺寸为0.71nm
×
0.71nm
×
1.82nm的近似圆柱形十二元环超笼,该超笼通过略微扭曲的十元环窗口(0.40nm
×
0.55nm)与外界连通。典型的mww型分子筛有mcm-22分子筛、itq-1、ssz-25、erb-1等。
5.专利cn104511271b公开了一种分子筛及其制备方法,通过引入特定的氮杂单环c
5-15
环烷烃(如n,n-二乙基环己基胺、二环己基胺、n-甲基二环己基胺等)和氮杂芳烃(如喹啉、n-苯基喹啉、n-环己基吡啶、6-甲基喹啉等)两种有机结构导向剂组合,以简便的方法制造出了具有mww结构的scm-1分子筛,该分子筛作为催化剂在苯与乙烯液相烷基化反应中具有优异的催化性能;该分子筛保留了位于层内的十元环孔道和相邻层结构之间的近似圆柱形十二元环超笼的十元环孔道,与mcm-22分子筛相比,该分子筛具有独特的“双”基础层结构,厚度在5nm左右,晶体更薄,因而表现出显著优于mcm-22分子筛的扩散性能。
6.含有杂原子的分子筛目前得到了许多研究人员的关注。将杂原子引入分子筛的骨架,可以使杂原子获得酸碱或氧化还原中心;同时杂原子原子半径较大,可以修饰分子筛的孔道,使得分子筛作为催化剂具有一些特殊且优异的性能,可以用于光电催化、离子交换、有机污染物的降解吸附及生物质转化制备高附加值的化学品等领域。
7.目前,杂原子分子筛的合成方法主要有直接水热合成法、离子交换法、浸渍法和二次合成法等。直接水热合成法是在反应过程中投入一些杂原子置换分子筛中的硅或
铝物种,通常在发生原子置换的过程中分子筛结构的规整度会降低。采用后处理的方法,在保持分子筛规整度的前提下,在含氟溶液中将一些杂原子如fe和ti置换分子筛中al的位置,得到了富含多种金属的高规整度杂原子分子筛,杂原子进入了分子筛的骨架形成四配位结
构,但是该处理方法需要使用含氟溶液,导致分子筛的后处理过程会产生较多的危险废弃物。cn 1552624a介绍了一种通过后处理对分子筛mcm-41进行改性的方法,但是金属离子分布不均匀且材料规整度降低。ti-mww分子筛一般采用采用含硼体系的水热合成法、无硼体系的双模板法及酸处理补钛等后处理方法。专利cn110203947a公开了一种钛硅分子筛ti-mww的制备方法,将mww分子筛、钛源和酸溶液混合经一定的处理在无氟体系下制得ti-mww分子筛,但该分子筛不含铝。掺杂不同杂原子的mww分子筛的制备方法也不同,因此,发展一种可以使杂原子有效引入mww分子筛骨架的普适性方法十分必要,对于杂原子催化剂的工业化应用具有积极的意义。
技术实现要素:
8.本发明所要解决的技术问题是提供一种可以使杂原子有效引入mww分子筛骨架,分散均匀,且该分子筛具有独特的化学组成和形貌,作为催化剂使用具有较好的性能。
9.本发明第一方面提供了一种分子筛,所述分子筛具有如式“msio2·
nal2o3·
pb2o3·
qmo
x”所示的示意性化学组成,其中25≤m/n≤500,m/p≥50,20≤m/q≤300,mo
x
为杂原子氧化物,且m元素进入分子筛骨架,x为m的化合价/2;所述分子筛包括质量分数为96~99.9%的mww结构分子筛,且厚度不高于15nm的片状晶体占统计总晶体数的至少80%。
10.进一步地,所述分子筛的示意性化学组成“msio2·
nal2o3·
pb2o3·
qmo
x”中,30≤m/n≤400,m/p≥100,25≤m/q≤250,优选30≤m/n≤300,m/p≥150,25≤m/q≤200;厚度不高于15nm的片状晶体占统计总晶体数的至少85%。
11.进一步地,所述杂原子氧化物mo
x
选自zro2、tio2、sno2、nb2o5、fe2o3、ga2o3、in2o3中的至少一种。
12.进一步地,所述分子筛的比表面积不低于380米2/克,优选为380~500米2/克;总孔容不低于0.45厘米3/克,优选为0.45~1.0厘米3/克,外比表面积不低于130米2/克,优选为130~200米2/克,外比表面积占总比表面积的比例不低于30%,优选为30%~50%。
13.进一步地,所述分子筛总酸量不小于400μmol/g,优选为400~1300μmol/g;总b酸量不小于150μmol/g,优选为150~450μmol/g;强b酸量不小于80μmol/g,优选为80~300μmol/g;总b酸量占总酸量不小于20%,优选为20~50%;强b酸量占总b酸量不小于30%,优选为30~70%。
14.进一步地,所述分子筛还包括质量分数为0.1~4%的其他结构材料magadiite。
15.本发明第二方面提供了一种分子筛的制备方法,包括以下步骤:
16.(1)将硅源、铝源、硼源、碱源、有机结构导向剂a、有机结构导向剂b和水混合,然后将上述混合物进行晶化处理,制得前驱体i;
17.(2)前驱体i经过铵离子交换后得到h型的前驱体i,再用水或水蒸气进行预处理,产物经分离、洗涤、干燥、焙烧制得前驱体ii;
18.(3)将前驱体ii与mo
x
杂原子氧化物前驱体研磨混合,产物经干燥、焙烧后得到分子筛。
19.进一步地,在步骤(1)中,所加入的硅源以sio2为计、铝源以al2o3为计、硼源以b2o3为计、碱源以oh-为计、有机结构导向剂a、有机结构导向剂b和水的摩尔配比为1:(0.002~0.04):(0.006~0.2):(0.05~0.4):(0.05~1.2):(0.005~0.25):(6~60),优选1:
(0.0025~0.033):(0.008~0.1):(0.065~0.35):(0.10~1.0):(0.01~0.2):(8~50),更优选1:(0.003~0.03):(0.01~0.05):(0.08~0.3):(0.15~0.8):(0.02~0.15):(10~40)。
20.进一步地,在步骤(1)中,所述硅源选自硅酸、硅胶、硅溶胶、硅酸四乙酯、水玻璃中的至少一种;所述铝源选自
氢氧化铝、氧化铝、铝酸盐、铝盐和四烷氧基铝中的至少一种;所述硼源选自硼酸、硼酸盐、硼砂和氧化硼中的至少一种。
21.进一步地,在步骤(1)中,所述碱源选自碱金属、碱土金属为阳离子的无机碱中的至少一种;所述有机结构导向剂a选自六亚甲基亚胺、哌啶、高哌嗪、环己胺、二环己胺、n,n-二乙基环己胺中的至少一种;所述有机结构导向剂b为n,n,n-三甲基金刚烷铵。
22.进一步地,在步骤(1)中,所述反应混合物的晶化条件为120~200℃晶化1~10天,优选130~190℃晶化1.5~9天,更优选140~180℃晶化2~8天。
23.进一步地,在步骤(1)中,所述晶化可以按照本领域常规已知的任何方式进行,比如可以举出使所述硅源、铝源、硼源、碱源、有机结构导向剂和水按照预定的比例混合,并使所获得的混合物在晶化条件下加热晶化的方法。
24.进一步地,在步骤(1)中,在晶化步骤结束之后,可以通过常规已知的任何分离方式和焙烧处理从所获得的混合物中得到产品。作为所述分离方式,比如可以举出对所述获得的混合物进行过滤、洗涤和干燥的方法。在此,所述过滤、洗涤和干燥可以按照本领域常规已知的任何方式进行。具体举例而言,作为所述过滤,比如可以简单地抽滤所述获得的产物混合物。作为所述洗涤,比如可以举出使用去离子水和/或乙醇进行洗涤。作为所述干燥温度,比如可以举出40~250℃,优选60~150℃,作为所述干燥的时间,比如可以举出8~30小时,优选10~20小时。该干燥可以在常压下进行,也可以在减压下进行。所述焙烧可以按照本领域常规已知的任何方式进行,比如焙烧温度一般为300~800℃,优选400~650℃,而焙烧时间一般为1~12小时,优选2~6小时。另外,所述焙烧一般在含氧气氛下进行,比如空气或者氧气气氛下。
25.进一步地,在步骤(2)中,所述前驱体i的铵离子交换为将前驱体i中的na
+
、k
+
等碱金属或碱土金属阳离子交换成nh
4+
,在20~60℃下交换0.5~4h,可以交换一次或多次,铵离子交换之后在60~120℃下干燥4~24h,再进行焙烧,焙烧温度为400~650℃,焙烧时间为1~12小时,焙烧气氛为氧气或空气,得到h型的前驱体i。
26.进一步地,在步骤(2)中,所述h型前驱体i在水中进行预处理的处理条件为40~100℃处理1小时~2天,优选50~90℃处理2小时~1.5天,更优选55~85℃处理4小时~1天;所述h型前驱体i与水的固液质量比为1:10~50。
27.进一步地,在步骤(2)中,所述h型前驱体i用水蒸气进行预处理的处理条件为200~800℃处理1~24小时,优选250~750℃处理2~21小时,更优选300~700℃处理3~18小时。
28.进一步地,在步骤(2)中,水或水蒸气预处理之后,分离、洗涤、干燥、焙烧采用常规方法,比如分离可采用离心分离的方法,洗涤可采用去离水进行洗涤,干燥可以在烘箱中进行。例如,干燥条件为:20~120℃下干燥2~24小时;焙烧条件为:焙烧温度为400~650℃,焙烧时间为1~12小时,焙烧气氛为氧气或空气。
29.进一步地,在步骤(3)中,所述mo
x
杂原子氧化物选自zro2、tio2、sno2、nb2o5、fe2o3、
ponndorf-verley reduction(mpv)反应制备环己醇具有较好的催化性能,反应物环己酮转化率》99%,产物环己醇选择性高达99%。
附图说明
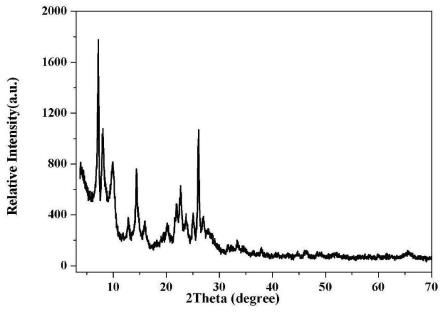
40.图1为实施例1中前驱体i焙烧前的x射线衍射谱图(xrd);
41.图2为实施例1中前驱体i的x射线衍射谱图(xrd);
42.图3为实施例1中前驱体i的扫描电子显微镜图(sem);
43.图4为实施例1中所得分子筛的x射线衍射谱图(xrd);
44.图5为实施例1中所得分子筛的扫描电子显微镜图(sem);
45.图6为实施例1中所得分子筛的x射线光电子能谱图(xps);
46.图7为实施例6中所得分子筛的x射线衍射谱图(xrd);
47.图8为实施例6中所得分子筛的扫描电子显微镜图(sem);
48.图9为实施例9中所得分子筛的x射线衍射谱图(xrd);
49.图10为实施例9中所得分子筛的x射线光电子能谱图(xps);
50.图11为实施例10中所得分子筛的x射线衍射谱图(xrd);
51.图12为实施例11中所得分子筛的x射线衍射谱图(xrd);
52.图13为实施例11中所得分子筛的扫描电子显微镜图(sem);
53.图14为实施例12中前驱体i焙烧前的x射线衍射谱图(xrd);
54.图15为实施例12中前驱体i的x射线衍射谱图(xrd);
55.图16为实施例12中前驱体i的扫描电子显微镜图(sem);
56.图17为实施例12中所得分子筛的x射线衍射谱图(xrd)。
具体实施方式
57.根据本发明,前述获得的分子筛可以以任何的物理形式应用,比如粉末状、颗粒状或者模制品状(比如条状、三叶草状等)。可以按照本领域常规已知的任何方式获得这些物理形式,并没有特别的限定。
58.在本说明书的上下文中,分子筛的结构是由x-射线衍射谱图(xrd)确定的,而x-射线衍射谱图(xrd)由x-射线粉末衍射仪测定,使用cu-kα射线源、
镍滤光片。样品测试前,采用扫描电子显微镜(sem)观察分子筛样品的结晶情况,确认样品中只含有一种晶体,即分子筛样品为纯相,在此基础上再进行xrd测试,确保xrd谱图中的衍射峰中没有其他晶体的干扰峰。
59.在本说明书的上下文中,包括在以下的实施例和对比例中,分子筛的x-射线粉末衍射仪的型号为panalytical x perpro型x-射线粉末衍射仪,分析样品的物相,cukα射线源镍滤光片,2θ扫描范围2~50
°
,操作电压40kv,电流40ma,扫描速率10
°
/min。
60.在本说明书的上下文中,包括在以下的实施例和对比例中,分子筛的扫描电子显微镜(sem)的型号为s-4800ii型场发射扫描电镜。
61.在本说明书的上下文中,包括在以下的实施例和对比例中,分子筛的晶体厚度的测量方法是:使用透射电子显微镜(荷兰fei公司g2f30透射电子显微镜,工作电压300kv)在10万倍的放大倍率下观测分子筛,随机选取一个观测视野,测量该观测视野中所有晶体的
厚度,统计厚度不高于15nm的晶体数占统计总晶体数的占比,重复该操作5次。以5次的平均值作为不高于15nm晶体数的占比。
62.根据本发明,前述获得的分子筛中的杂原子m有效地进入了分子筛的骨架中。可以根据xps(x射线光电子能谱分析)测试分子筛表面元素结合能或紫外拉曼判断杂原子m的配位的状态。
63.在本说明书的上下文中,包括在以下的实施例和对比例中,分子筛的x射线光电子能谱仪(xps)的型号为thermo公司的esca lab-250型x射线光电子能谱仪,采用c1s=284.6ev为内标校正所测元素信号。
64.本发明采用简单的研磨方法,将基本脱硼和部分脱铝后的前驱体与mo
x
杂原子氧化物前驱体研磨,所得分子筛再无需进行铵离子交换,直接焙烧即可作为催化剂使用;该方法具有普适性,可以有效将杂原子引入分子筛的骨架。
65.在本说明书的上下文中,包括在以下的实施例和对比例中,分子筛的孔容、比表面积、外比表面积是通过氮气物理吸脱附法(bet法)测得的:利用物理吸附仪(micromeretic asap2020m物理吸附仪)测得分子筛的氮气物理吸脱附等温线,再经bet方程式和t-plot方程式进行计算。针对该分子筛的实验条件为:测量温度-169℃,测量前先将分子筛在550℃空气气氛下热处理6小时,再在350℃真空预处理4小时。
66.在本说明书的上下文中,包括在以下的实施例和对比例中,分子筛的电感耦合等离子体原子发射光谱仪(icp)型号为varian 725-es,将分析样品用氢氟酸溶解检测得到元素的含量,以摩尔计。
67.在本说明书的上下文中,包括在以下的实施例和对比例中,采用吡啶吸附红外方法(nicolet model 710光谱仪)对分子筛酸量、酸种类进行测定。具体操作步骤如下:a、样品预处理。将样品(约30mg)压片成型为直径13mm的薄圆片,并装入红外样品槽中。之后,样品在真空池条件和400℃下预处理1h。待样品槽冷却至室温,扫描样品红外数据作为背景。b、吡啶吸附。在室温下和真空环境下,将吡啶蒸气通入至原位直至吸附达到平衡,吸附时间为1h。c、吡啶脱附。吸附结束后,在100℃下抽真空至内部压力不再变化,脱附时间为40min,并分别扫描记录红外吸收光谱。吡啶吸附前后的差谱即为所得的吡啶吸附-红外吸收光谱图。根据图谱对样品的酸量进行了半定量计算:
[0068][0069]
其中r和w为催化剂薄圆片的直径(cm)和质量(g),a为根据扫描吡啶吸附-红外吸收光谱图在指定波数峰的吸光度积分数值。imec为积分摩尔消光系数,imec
l
为2.22,imecb为1.67。1545cm-1
附近的峰为b酸,1455cm-1
附近的峰为l酸,其中400℃下吡啶脱附后得到b酸的峰可以认为是强b酸,根据图谱对应的酸量为强b酸的酸量。
[0070]
反应产物环己醇用气质联用(gc-ms)分析定性和定量,用高效液相色谱(hplc)分析产物可溶性碳水化合物的转化率。气质联用仪为美国安捷伦公司的agilent 7890a,色谱柱为hp-5非极性毛细管柱(30m,0.53mm),气相色谱仪为agilent 7890b,检测器为氢焰离子化检测器(fid),色谱柱为se-54毛细管柱(30m,0.53mm)。高效液相色谱采用agilent 1200型分析,色谱柱为shodex sc1011糖柱(8
×
300mm)。
[0071]
以下采用实施例进一步详细地说明本发明,但本发明并不限于这些实施例。
[0072]
实施例1
[0073]
将628.44克去离子水、14.26克铝酸钠(含al2o
3 43.0重量%,na2o 35.0重量%)、4.39克氢氧化钠、89.47克六亚甲基亚胺(有机结构导向剂(a))、101.21克n,n,n-三甲基金刚烷铵溶液(含n,n,n-三甲基金刚烷铵25.12重量%)(有机结构导向剂(b))、4.65克硼酸和451.70克硅溶胶(含sio
2 40.0重量%),室温搅拌2小时后制得混合物,最终物料配比(摩尔比)为:
[0074]
sio2/al2o3=50;
[0075]
sio2/b2o3=40;
[0076]
naoh/sio2=0.09;
[0077]
六亚甲基亚胺/sio2=0.30;
[0078]
n,n,n-三甲基金刚烷铵/sio2=0.04;
[0079]
h2o/sio2=18。
[0080]
混合物装入不锈钢反应釜中,在150℃下搅拌加热晶化5天。晶化结束后过滤、洗涤,在100℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体i。
[0081]
将前驱体i与0.2mol/l的nh4no3溶液(质量比1:20)在45℃下进行铵离子交换2小时,然后离心洗涤,重复铵离子交换两次后得到的样品在100℃下过夜烘干,在550℃空气中焙烧6小时制得h型的前驱体i。将h型的前驱体i(固)加入水中,固液质量比为1:20,混合物在65℃水浴搅拌条件下预处理16个小时;产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0082]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:20,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0083]
焙烧前的前驱体i的xrd谱图如图1所示,为mww分子筛和magadiite的混合物,其中magadiite的质量含量为2.0%,焙烧后得到的前驱体i的xrd谱图如图2所示,主要为mww结构的分子筛,分子筛的质量含量为99.0%,前驱体i的sem电镜图如图3所示。
[0084]
产品xrd谱图如图4所示,为mww结构的分子筛,其质量含量为99.1%,magadiite的质量含量为0.9%;样品的sem电镜图如图5所示,晶体呈纳米片状,85%的晶体厚度小于10nm;样品的xps谱图如图6所示,说明zr成功进入了mww分子筛的骨架。
[0085]
样品的比表面积423米2/克;总孔容0.68厘米3/克,经bet法测得的外比表面积155米2/克,外比表面积占总比表面积的比例为37%。
[0086]
采用电感耦合等离子体原子发射光谱(icp)测得前驱体i的sio2/al2o3摩尔比为52.6,sio2/b2o3摩尔比为39.6;h型前驱体i经吡啶吸附红外py-ir该分子筛的总酸量为711μmol/g。
[0087]
采用电感耦合等离子体原子发射光谱(icp)测得zr-mww分子筛的sio2/al2o3摩尔比为57.1,sio2/zro2摩尔比为47.3,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0088]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为683μmol/g,总b酸量为249μmol/g,强b酸量为133μmol/g,总b酸量占总酸量为36%,强b酸量占总b酸量为53%。最终得到的分子筛的总酸量大于h型前驱体i的90%。
[0089]
实施例2
[0090]
同实施例1,制得h型的前驱体i。
[0091]
将h型的前驱体i(固)加入水中,固液质量比为1:20,混合物在65℃水浴搅拌条件下预处理24个小时;产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0092]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:30,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0093]
产品xrd谱图与图4类似,为mww结构的分子筛,其质量含量为99.2%,magadiite的质量含量为0.8%;样品的sem电镜图与图5类似,晶体呈纳米片状,85%的晶体厚度小于12nm;样品的xps谱图与图6类似,zr进入了mww分子筛的骨架。
[0094]
样品的比表面积441米2/克;总孔容0.62厘米3/克,经bet法测得的外比表面积139米2/克,外比表面积占总比表面积的比例为32%。
[0095]
采用电感耦合等离子体原子发射光谱(icp)测得zr-mww分子筛的sio2/al2o3摩尔比为59.8,sio2/zro2摩尔比为67.6,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0096]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为622μmol/g,总b酸量为232μmol/g,强b酸量为124μmol/g,总b酸量占总酸量为37%,强b酸量占总b酸量为53%。最终得到的分子筛的总酸量大于h型前驱体i的85%。
[0097]
实施例3
[0098]
同实施例1,制得h型的前驱体i。
[0099]
将h型的前驱体i(固)加入水中,固液质量比为1:20,混合物在80℃水浴搅拌条件下预处理12个小时。产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0100]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:25,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0101]
样品xrd谱图与图4类似,为mww结构的分子筛,其质量含量为99.3%,magadiite的质量含量为0.7%;样品的sem图与图5类似,晶体呈纳米片状,88%的晶体厚度小于10nm;样品的xps谱图与图6类似,zr进入了mww分子筛的骨架。
[0102]
样品的比表面积396米2/克;总孔容0.58厘米3/克,经bet法测得的外比表面积172米2/克,外比表面积占总比表面积的比例为43%。
[0103]
采用电感耦合等离子体原子发射光谱(icp)测得样品的sio2/al2o3摩尔比为56.9,sio2/zro2摩尔比为58.9,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0104]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为654μmol/g,总b酸量为227μmol/g,强b酸量为118μmol/g,总b酸量占总酸量为35%,强b酸量占总b酸量为52%。最终得到的分子筛的总酸量大于h型前驱体i的90%。
[0105]
实施例4
[0106]
同实施例1,制得h型的前驱体i。
[0107]
将h型的前驱体i(固)加入水中,固液质量比为1:20,混合物在80℃水浴搅拌条件下预处理24个小时;产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0108]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:15,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0109]
产品xrd谱图与图4类似,为mww结构的分子筛,其质量含量为99.2%,magadiite的质量含量为0.8%;样品的sem电镜图与图5类似,晶体呈纳米片状,85%的晶体厚度小于12nm;样品的xps谱图与图6类似,zr进入了mww分子筛的骨架。
[0110]
样品的比表面积408米2/克;总孔容0.64厘米3/克,经bet法测得的外比表面积164米2/克,外比表面积占总比表面积的比例为40%。
[0111]
采用电感耦合等离子体原子发射光谱(icp)测得zr-mww分子筛的sio2/al2o3摩尔比为62.4,sio2/zro2摩尔比为45.1,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0112]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为623μmol/g,总b酸量为214μmol/g,强b酸量为115μmol/g,总b酸量占总酸量为34%,强b酸量占总b酸量为54%。最终得到的分子筛的总酸量大于h型前驱体i的85%。
[0113]
实施例5
[0114]
同实施例1,制得h型的前驱体i。
[0115]
将h型的前驱体i(固)加入水中,固液质量比为1:20,混合物在55℃水浴搅拌条件下预处理24个小时。产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0116]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:40,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0117]
样品xrd谱图与图4类似,为mww结构的分子筛,其质量含量为99.2%,magadiite的质量含量为0.8%;样品的sem图与图5类似,晶体呈纳米片状,90%的晶体厚度小于12nm;样品的xps谱图与图6类似,zr进入了mww分子筛的骨架。
[0118]
样品的比表面积448米2/克;总孔容0.61厘米3/克,经bet法测得的外比表面积166米2/克,外比表面积占总比表面积的比例为37%。
[0119]
采用电感耦合等离子体原子发射光谱(icp)测得样品的sio2/al2o3摩尔比为56.2,sio2/zro2摩尔比为87.7,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0120]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为619μmol/g,总b酸量为242μmol/g,强b酸量为142μmol/g,总b酸量占总酸量为39%,强b酸量占总b酸量为59%。最终得到的分子筛的总酸量大于h型前驱体i的85%。
[0121]
实施例6
[0122]
同实施例1,制得h型的前驱体i。
[0123]
将h型的前驱体i放入管式炉中,将管式炉以10℃/min的速度升至500℃,然后通入纯水蒸气,在此条件下预处理4个小时,然后降至室温。产物离心洗涤两次,110℃下干燥过
夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0124]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:20,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0125]
样品xrd谱图如图7所示,为mww结构的分子筛,其质量含量为99.3%,magadiite的质量含量为0.7%;样品的sem图与如图8所示,晶体呈纳米片状,90%的晶体厚度小于12nm;样品的xps谱图与图6类似,zr进入了mww分子筛的骨架。
[0126]
样品的比表面积422米2/克;总孔容0.63厘米3/克,经bet法测得的外比表面积145米2/克,外比表面积占总比表面积的比例为34%。
[0127]
采用电感耦合等离子体原子发射光谱(icp)测得样品的sio2/al2o3摩尔比为55.6,sio2/zro2摩尔比为46.1,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0128]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为701μmol/g,总b酸量为281μmol/g,强b酸量为136μmol/g,总b酸量占总酸量为40%,强b酸量占总b酸量为48%。最终得到的分子筛的总酸量大于h型前驱体i的95%。
[0129]
实施例7
[0130]
同实施例1,制得h型的前驱体i。
[0131]
将h型的前驱体i放入管式炉中,将管式炉以10℃/min的速度升至550℃,然后通入纯水蒸气,在此条件下预处理4个小时,然后降至室温。产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0132]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:30,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0133]
样品xrd谱图与图4类似,为mww结构的分子筛,其质量含量为99.4%,magadiite的质量含量为0.6%;样品的sem图与图5类似,晶体呈纳米片状,85%的晶体厚度小于10nm;样品的xps谱图与图6类似,zr进入了mww分子筛的骨架。
[0134]
样品的比表面积417米2/克;总孔容0.67厘米3/克,经bet法测得的外比表面积157米2/克,外比表面积占总比表面积的比例为38%。
[0135]
采用电感耦合等离子体原子发射光谱(icp)测得样品的sio2/al2o3摩尔比为58.9,sio2/zro2摩尔比为69.7,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0136]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为623μmol/g,总b酸量为249μmol/g,强b酸量为129μmol/g,总b酸量占总酸量为40%,强b酸量占总b酸量为52%。最终得到的分子筛的总酸量大于h型前驱体i的85%。
[0137]
实施例8
[0138]
同实施例1,制得h型的前驱体i。
[0139]
将h型的前驱体i放入管式炉中,将管式炉以10℃/min的速度升至650℃,然后通入纯水蒸气,在此条件下预处理2个小时,然后降至室温。产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0140]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:20,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘
箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0141]
样品xrd谱图与图4类似,为mww结构的分子筛,其质量含量为99.3%,magadiite的质量含量为0.7%;样品的sem图与图5类似,晶体呈纳米片状,85%的晶体厚度小于10nm;样品的xps谱图与图6类似,zr进入了mww分子筛的骨架。
[0142]
样品的比表面积429米2/克;总孔容0.60厘米3/克,经bet法测得的外比表面积148米2/克,外比表面积占总比表面积的比例为35%。
[0143]
采用电感耦合等离子体原子发射光谱(icp)测得样品的sio2/al2o3摩尔比为60.3,sio2/zro2摩尔比为46.0,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0144]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为660μmol/g,总b酸量为236μmol/g,强b酸量为98μmol/g,总b酸量占总酸量为36%,强b酸量占总b酸量为42%。最终得到的分子筛的总酸量大于h型前驱体i的90%。
[0145]
实施例9
[0146]
同实施例4,制得前驱体ii。
[0147]
将前驱体ii和sno2前驱体(ch3)2sncl2倒入研钵中,sn((ch3)2sncl2中sn的理论量)与前驱体ii的质量比为1:20,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品sn-mww分子筛。
[0148]
样品xrd谱图如图9所示,为mww结构的分子筛,其质量含量为99.3%,magadiite的质量含量为0.7%;样品的sem图与图5类似,晶体呈纳米片状,92%的晶体厚度小于10nm;样品的xps谱图如图10所示,sn进入了mww分子筛的骨架。
[0149]
样品的比表面积456米2/克;总孔容0.61厘米3/克,经bet法测得的外比表面积148米2/克,外比表面积占总比表面积的比例为33%。
[0150]
采用电感耦合等离子体原子发射光谱(icp)测得样品的sio2/al2o3摩尔比为61.9,sio2/sno2摩尔比为52.5,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0151]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为627μmol/g,总b酸量为221mol/g,强b酸量为119μmol/g,总b酸量占总酸量为35%,强b酸量占总b酸量为54%。最终得到的分子筛的总酸量大于h型前驱体i的85%。
[0152]
实施例10
[0153]
同实施例7,制得前驱体ii。
[0154]
将前驱体ii和tio2前驱体cp2ticl2倒入研钵中,ti(cp2ticl2中ti的理论量)与前驱体ii的质量比为1:50,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品ti-mww分子筛。
[0155]
样品xrd谱图如图11所示,为mww结构的分子筛,其质量含量为99.3%,magadiite的质量含量为0.7%;样品的sem图与图5类似,晶体呈纳米片状,88%的晶体厚度小于10nm;样品的xps测试表明ti进入了mww分子筛的骨架。
[0156]
样品的比表面积433米2/克;总孔容0.55厘米3/克,经bet法测得的外比表面积162米2/克,外比表面积占总比表面积的比例为37%。
[0157]
采用电感耦合等离子体原子发射光谱(icp)测得样品的sio2/al2o3摩尔比为58.1,sio2/tio2摩尔比为106.6,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0158]
样品经吡啶吸附红外py-ir该分子筛的总酸量为598μmol/g,总b酸量为210μmol/g,强b酸量为121μmol/g,总b酸量占总酸量为35%,强b酸量占总b酸量为58%。最终得到的分子筛的总酸量大于h型前驱体i的80%。
[0159]
实施例11
[0160]
将45.71克去离子水、0.813克铝酸钠(含al2o
3 43.0重量%,na2o 35.0重量%)、0.73克氢氧化钠、6.80克六亚甲基亚胺(有机结构导向剂(a))、11.54克n,n,n-三甲基金刚烷铵溶液(含n,n,n-三甲基金刚烷铵25.12重量%)(有机结构导向剂(b))、0.565克硼酸和41.20克硅溶胶(含sio
2 40.0重量%),室温搅拌2小时后制得混合物,最终物料配比(摩尔比)为:
[0161]
sio2/al2o3=80;
[0162]
sio2/b2o3=30;
[0163]
naoh/sio2=0.10;
[0164]
六亚甲基亚胺/sio2=0.25;
[0165]
n,n,n-三甲基金刚烷铵/sio2=0.05;
[0166]
h2o/sio2=16。
[0167]
混合物装入不锈钢反应釜中,在150℃下搅拌加热晶化3天。晶化结束后过滤、洗涤,在100℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体i。
[0168]
将前驱体i与0.2mol/l的nh4no3溶液(质量比1:20)在45℃下进行铵离子交换2小时,然后离心洗涤,重复铵离子交换两次后得到的样品在100℃下过夜烘干,在550℃空气中焙烧6小时制得h型的前驱体i。将h型的前驱体i(固)加入水中,固液质量比为1:20,混合物在60℃水浴搅拌条件下预处理16个小时;产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0169]
将前驱体ii和sno2前驱体(ch3)2sncl2倒入研钵中,sn((ch3)2sncl2中sn的理论量)与前驱体ii的质量比为1:20,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品sn-mww分子筛。
[0170]
焙烧前的前驱体i的xrd谱图与图1类似,为mww分子筛和magadiite的混合物,其中magadiite的质量含量小于2%;焙烧后得到的前驱体i的xrd谱图与图2类似,主要为mww结构的分子筛,分子筛的质量含量为98.5%,前驱体i的sem电镜图与图3类似。
[0171]
样品xrd谱图如图12所示,为mww结构的分子筛,其质量含量为99.0%,magadiite的质量含量为1.0%;样品的sem图如图13所示,晶体呈纳米片状,92%的晶体厚度小于10nm;样品的xps谱图与图10类似,sn进入了mww分子筛的骨架。
[0172]
样品的比表面积420米2/克;总孔容0.65厘米3/克,经bet法测得的外比表面积149米2/克,外比表面积占总比表面积的比例为37%。
[0173]
采用电感耦合等离子体原子发射光谱(icp)测得前驱体i的sio2/al2o3摩尔比为81.6,sio2/b2o3摩尔比为30.5。样品经吡啶吸附红外py-ir该分子筛的总酸量为560μmol/g。
[0174]
采用电感耦合等离子体原子发射光谱(icp)测得sn-mww分子筛的sio2/al2o3摩尔比为86.7,sio2/sno2摩尔比为53.7,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<20%。
[0175]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为485μmol/g,总
b酸量为211μmol/g,强b酸量为113μmol/g,总b酸量占总酸量为44%,强b酸量占总b酸量为54%。最终得到的分子筛的总酸量大于h型前驱体i的85%。
[0176]
实施例12
[0177]
将25.11克去离子水、0.387克铝酸钠(含al2o
3 43.0重量%,na2o 35.0重量%)、0.412克氢氧化钠、5.66克六亚甲基亚胺(有机结构导向剂(a))、9.60克n,n,n-三甲基金刚烷铵溶液(含n,n,n-三甲基金刚烷铵25.12重量%)(有机结构导向剂(b))、0.403克硼酸和24.49克硅溶胶(含sio
2 40.0重量%),室温搅拌2小时后制得混合物,最终物料配比(摩尔比)为:
[0178]
sio2/al2o3=100;
[0179]
sio2/b2o3=25;
[0180]
naoh/sio2=0.09;
[0181]
六亚甲基亚胺/sio2=0.35;
[0182]
n,n,n-三甲基金刚烷铵/sio2=0.07;
[0183]
h2o/sio2=16。
[0184]
混合物装入不锈钢反应釜中,在150℃下搅拌加热晶化4天。晶化结束后过滤、洗涤,在100℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体i。
[0185]
将前驱体i与0.2mol/l的nh4no3溶液(质量比1:20)在45℃下进行铵离子交换2小时,然后离心洗涤,重复铵离子交换两次后得到的样品在100℃下过夜烘干,在550℃空气中焙烧6小时制得h型的前驱体i。将h型的前驱体i放入管式炉中,将管式炉以10℃/min的速度升至400℃,然后通入纯水蒸气,在此条件下预处理6个小时,然后降至室温。产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0186]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:40,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0187]
焙烧前的前驱体i的xrd谱图如图14所示,为mww分子筛和magadiite的混合物,其中magadiite的质量含量为18%;焙烧后得到的前驱体i的xrd谱图如图15所示,主要为mww结构的分子筛,分子筛的质量含量为98.0%,前驱体i的sem电镜图如图16所示。
[0188]
产品xrd谱图如图17所示,为mww结构的分子筛,其质量含量为98.5%,magadiite的质量含量为1.5%;样品的sem电镜图与图16类似,晶体呈纳米片状,85%的晶体厚度小于10nm;样品的xps谱图与图6类似,zr成功进入了mww分子筛的骨架。
[0189]
样品的比表面积445米2/克;总孔容0.66厘米3/克,经bet法测得的外比表面积152米2/克,外比表面积占总比表面积的比例为34%。
[0190]
采用电感耦合等离子体原子发射光谱(icp)测得前驱体i的sio2/al2o3摩尔比为102.1,sio2/b2o3摩尔比为24.4。h型的前驱体i经吡啶吸附红外py-ir该分子筛的总酸量为480μmol/g。
[0191]
采用电感耦合等离子体原子发射光谱(icp)测得zr-mww分子筛的sio2/al2o3摩尔比为103.5,sio2/zro2摩尔比为85.6,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<10%。
[0192]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为422μmol/g,总
b酸量为203μmol/g,强b酸量为103μmol/g,总b酸量占总酸量为48%,强b酸量占总b酸量为51%。最终得到的分子筛的总酸量大于h型前驱体i的85%。
[0193]
实施例13
[0194]
将43.16克去离子水、1.585克铝酸钠(含al2o
3 43.0重量%,na2o 35.0重量%)、0.086克氢氧化钠、5.96克六亚甲基亚胺(有机结构导向剂(a))、5.06克n,n,n-三甲基金刚烷铵溶液(含n,n,n-三甲基金刚烷铵25.12重量%)(有机结构导向剂(b))、0.496克硼酸和30.11克硅溶胶(含sio
2 40.0重量%),室温搅拌2小时后制得混合物,最终物料配比(摩尔比)为:
[0195]
sio2/al2o3=30;
[0196]
sio2/b2o3=25;
[0197]
naoh/sio2=0.10;
[0198]
六亚甲基亚胺/sio2=0.30;
[0199]
n,n,n-三甲基金刚烷铵/sio2=0.03;
[0200]
h2o/sio2=18。
[0201]
混合物装入不锈钢反应釜中,在150℃下搅拌加热晶化5天。晶化结束后过滤、洗涤,在100℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体i。
[0202]
将前驱体i与0.2mol/l的nh4no3溶液(质量比1:20)在45℃下进行铵离子交换2小时,然后离心洗涤,重复铵离子交换两次后得到的样品在100℃下过夜烘干,在550℃空气中焙烧6小时制得h型的前驱体i。将h型的前驱体i放入管式炉中,将管式炉以10℃/min的速度升至600℃,然后通入纯水蒸气,在此条件下预处理2个小时,然后降至室温。产物离心洗涤两次,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0203]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:30,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0204]
焙烧前的前驱体i的xrd谱图与图1类似,为mww分子筛和magadiite的混合物,其中magadiite的质量含量小于1%;焙烧后得到的前驱体i的xrd谱图与图2类似,主要为mww结构的分子筛,分子筛的质量含量为99.2%,前驱体i的sem电镜图与图3类似。
[0205]
产品xrd谱图与图4类似,为mww结构的分子筛,分子筛的质量含量为99.5%,magadiite的质量含量为0.5%;样品的sem电镜图与图5类似,晶体呈纳米片状,85%的晶体厚度小于10nm;样品的xps谱图与图6类似,zr成功进入了mww分子筛的骨架。
[0206]
样品的比表面积430米2/克;总孔容0.69厘米3/克,经bet法测得的外比表面积146米2/克,外比表面积占总比表面积的比例为34%。
[0207]
采用电感耦合等离子体原子发射光谱(icp)测得前驱体i的sio2/al2o3摩尔比为31.2,sio2/b2o3摩尔比为25.3。h型的前驱体样品经吡啶吸附红外py-ir该分子筛的总酸量为1133μmol/g。
[0208]
采用电感耦合等离子体原子发射光谱(icp)测得zr-mww分子筛的sio2/al2o3摩尔比为32.9,sio2/zro2摩尔比为69.1,sio2/b2o3摩尔比大于200;硼的脱除率>80%,铝的脱除率<10%。
[0209]
最终得到的分子筛样品经吡啶吸附红外py-ir该分子筛的总酸量为1053μmol/g,
总b酸量为384μmol/g,强b酸量为196μmol/g,总b酸量占总酸量为36%,强b酸量占总b酸量为51%。最终得到的分子筛的总酸量大于h型前驱体i的90%。
[0210]
对比例1
[0211]
同实施例5,制得h型的前驱体i。
[0212]
将h型的前驱体i(固)加入到浓度为3mol/l的h2c2o4溶液(液)中,固液比(质量)为1:20,混合物在65℃水浴搅拌条件下预处理16个小时。产物离心洗涤至溶液ph=7,110℃下干燥过夜,然后在550℃空气中焙烧6小时,制得前驱体ii。
[0213]
将前驱体ii和zro2前驱体cp2zrcl2倒入研钵中,zr(cp2zrcl2中zr的理论量)与前驱体ii的质量比为1:20,混合物在研钵中充分研磨得到分散均匀的混合物;混合物在110℃烘箱中干燥过夜,然后在550℃空气中焙烧6小时得到产品zr-mww分子筛。
[0214]
样品xrd谱图与图4类似,为mww结构的分子筛,magadiite的质量含量为0;样品的sem图与图5类似,晶体呈纳米片状,90%的晶体厚度小于12nm;样品的xps谱图与图6类似,zr进入了mww分子筛的骨架。
[0215]
样品的比表面积388米2/克;总孔容0.79厘米3/克,经bet法测得的外比表面积128米2/克,外比表面积占总比表面积的比例为30%。
[0216]
采用电感耦合等离子体原子发射光谱(icp)测得样品的sio2/al2o3摩尔比为129.2,sio2/zro2摩尔比为47.7,sio2/b2o3摩尔比大于500;硼的脱除率>90%,铝的脱除率>60%。
[0217]
样品经吡啶吸附红外py-ir该分子筛的总酸量为306μmol/g,总b酸量为89μmol/g,强b酸量为52μmol/g,总b酸量占总酸量为29%,强b酸量占总b酸量为58%。最终得到的分子筛的总酸量小于h型前驱体i的45%。
[0218]
实施例14~21
[0219]
分别按照实施例1~8的方法得到的分子筛用作催化剂,用于环己酮和异丙醇的meerwein-ponndorf-verley reduction(mpv)反应制备环己醇。
[0220]
将0.1g上述制备的催化剂、1.0mmol环己酮和60mmol异丙醇加入带搅拌的高压反应釜中。采用程序升温加热套升至预设温度,并采用磁力搅拌然后搅拌。反应条件、催化剂活性和产物选择性见表1。
[0221]
对比例2
[0222]
按照对比例1方法得到的分子筛用作催化剂,用于环己酮和异丙醇的meerwein-ponndorf-verley reduction(mpv)反应制备环己醇。
[0223]
将0.1g上述制备的催化剂、1.0mmol环己酮和60mmol异丙醇加入带搅拌的高压反应釜中。采用程序升温加热套升至预设温度,并采用磁力搅拌然后搅拌。反应条件、催化剂活性和产物选择性见表1。
[0224]
表1 实施例14~21和对比例2催化剂性能结果
[0225][0226]
以上详细描述了本发明的具体实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。技术特征:
1.一种分子筛,所述分子筛具有如式“msio2·
nal2o3·
pb2o3·
qmo
x”所示的示意性化学组成,其中25≤m/n≤500,m/p≥50,20≤m/q≤300,mo
x
为杂原子氧化物,且m元素进入分子筛骨架,x为m的化合价/2;所述分子筛包括质量分数为96~99.9%的mww结构分子筛,且厚度不高于15nm的片状晶体占统计总晶体数的至少80%。2.按照权利要1所述的分子筛,其特征在于,所述分子筛的示意性化学组成“msio2·
nal2o3·
pb2o3·
qmo
x”中,30≤m/n≤400,m/p≥100,25≤m/q≤250,优选30≤m/n≤300,m/p≥150,25≤m/q≤200;厚度不高于15nm的片状晶体占统计总晶体数的至少85%。3.按照权利要1所述的分子筛,其特征在于,所述杂原子氧化物mo
x
选自zro2、tio2、sno2、nb2o5、fe2o3、ga2o3、in2o3中的至少一种。4.按照权利要求1所述的分子筛,其特征在于,所述分子筛的比表面积不低于380米2/克,优选为380~500米2/克;总孔容不低于0.45厘米3/克,优选为0.45~1.0厘米3/克,外比表面积不低于130米2/克,优选为130~200米2/克,外比表面积占总比表面积的比例不低于30%,优选为30%~50%;和/或,所述分子筛总酸量不小于400μmol/g,优选为400~1300μmol/g;总b酸量不小于150μmol/g,优选为150~450μmol/g;强b酸量不小于80μmol/g,优选为80~300μmol/g;总b酸量占总酸量不小于20%,优选为20~50%;强b酸量占总b酸量不小于30%,优选为30~70%。5.按照权利要求1-4任一所述的分子筛,其特征在于,所述分子筛中还包括质量分数为0.1~4%的其他结构材料magadiite。6.一种分子筛的制备方法,包括以下步骤:(1)将硅源、铝源、硼源、碱源、有机结构导向剂a、有机结构导向剂b和水混合,然后将上述混合物进行晶化处理,制得前驱体i;(2)前驱体i经过铵离子交换后得到h型前驱体i,再用水或水蒸气进行预处理,产物经分离、洗涤、干燥、焙烧制得前驱体ii;(3)将前驱体ii与mo
x
杂原子氧化物前驱体研磨混合,产物经干燥、焙烧后得到分子筛。7.按照权利要求6所述的制备方法,其特征在于,在步骤(1)中,所加入的硅源以sio2为计、铝源以al2o3为计、硼源以b2o3为计、碱源以oh-为计、有机结构导向剂a、有机结构导向剂b和水的摩尔配比为1:(0.002~0.04):(0.006~0.2):(0.05~0.4):(0.05~1.2):(0.005~0.25):(6~60),优选1:(0.0025~0.033):(0.008~0.1):(0.065~0.35):(0.10~1.0):(0.01~0.2):(8~50),更优选1:(0.003~0.03):(0.01~0.05):(0.08~0.3):(0.15~0.8):(0.02~0.15):(10~40)。8.按照权利要求6所述的制备方法,其特征在于,在步骤(1)中,所述硅源选自硅酸、硅胶、硅溶胶、硅酸四乙酯、水玻璃中的至少一种;所述铝源选自氢氧化铝、氧化铝、铝酸盐、铝盐和四烷氧基铝中的至少一种;所述硼源选自硼酸、硼酸盐、硼砂和氧化硼中的至少一种;和/或在步骤(1)中,所述碱源选自碱金属、碱土金属为阳离子的无机碱中的至少一种;所述有机结构导向剂a选自六亚甲基亚胺、哌啶、高哌嗪、环己胺、二环己胺、n,n-二乙基环己胺中的至少一种;所述有机结构导向剂b为n,n,n-三甲基金刚烷铵;和/或,在步骤(3)中,mo
x
杂原子氧化物选自zro2、tio2、sno2、nb2o5、fe2o3、ga2o3、in2o3中的至少一种;所述zro2前驱体选自含锆有机金属配合物、锆盐、二氧化锆中的至少一种,优
选含锆有机金属配合物;所述tio2前驱体选自含钛有机金属配合物、钛盐、钛酸四丁酯、二氧化钛中的至少一种,优选含钛有机金属配合物;所述sno2前驱体选自含
锡有机金属配合物、锡盐、二氧化锡中的至少一种,优选含锡有机金属配合物;所述nb2o5前驱体选自含铌有机金属配合物、铌盐中的至少一种,优选含铌有机金属配合物;所述fe2o3前驱体选自含铁有机金属配合物、铁盐中的至少一种,优选含铁有机金属配合物;所述ga2o3前驱体选自含镓有机金属配合物、镓盐中的至少一种,优选含镓有机金属配合物;所述in2o3前驱体选自含铟有机金属配合物、铟盐中的至少一种,优选含铟有机金属配合物。9.按照权利要求6所述的制备方法,其特征在于,在步骤(1)中,所述反应混合物的晶化条件为120~200℃晶化1~10天,优选130~190℃晶化1.5~9天,更优选140~180℃晶化2~8天。10.按照权利要求6所述的制备方法,其特征在于,在步骤(2)中,所述h型前驱体i在水中进行预处理的处理条件为40~100℃处理1小时~2天,优选50~90℃处理2小时~1.5天,更优选55~85℃处理4小时~1天;所述h型前驱体i与水的固液质量比为1:10~50;或者,在步骤(2)中,所述h型前驱体i用水蒸气进行预处理的处理条件为200~800℃处理1~24小时,优选250~750℃处理2~21小时,更优选300~700℃处理3~18小时。11.按照权利要求6~10任一项所述的分子筛的制备方法,其特征在于,所述的前驱体i在晶化处理步骤中干燥后,焙烧前的形态为质量分数为60~85%的mww结构、1~20%的magadiite材料、5~15%的有机结构导向剂和1~5%水;所述的前驱体i经过晶化处理中的焙烧后的形态为质量分数95~99.9%的mww结构和0.1~5%的magadiite材料。12.按照权利要求6~10任一项所述分子筛的制备方法,其特征在于,所述制备方法中得到的分子筛中硼的脱除率>80%,铝的脱除率<20%。13.按照权利要求6~10任一项所述分子筛的制备方法,其特征在于,所述制备方法中得到的分子筛的总酸量不低于步骤(2)中的h型前驱体i的80%。14.一种分子筛组合物,包含按照权利要求1~5任一所述的分子筛或者按照权利要求6~13任一所述的制备方法制备的分子筛,以及粘结剂。15.按照权利要求1~5任一所述的分子筛或者按照权利要求6~13任一所述的制备方法制备的分子筛、或者按照权利要求14所述的分子筛组合物作为有机化合物转化用催化剂的应用。
技术总结
本发明公开了一种分子筛及其制备方法和应用。所述分子筛具有如式“mSiO2·
技术研发人员:刘闯 王振东 李相呈 李月坤 马多征 梁俊
受保护的技术使用者:中国石油化工股份有限公司上海石油化工研究院
技术研发日:2021.05.19
技术公布日:2022/11/24
声明:
“分子筛及其制备方法和应用与流程” 该技术专利(论文)所有权利归属于技术(论文)所有人。仅供学习研究,如用于商业用途,请联系该技术所有人。
我是此专利(论文)的发明人(作者)
 740
编辑:北方有色网
来源:中国石油化工股份有限公司上海石油化工研究院
740
编辑:北方有色网
来源:中国石油化工股份有限公司上海石油化工研究院



 咨询细节
咨询细节











































































 2026年08月06日 ~ 08日
2026年08月06日 ~ 08日 















