本公开属于生物医用材料领域,涉及硅磷酸钙基体粉料及制备方法、骨修复材料及制备方法。
背景技术:
:这里的陈述仅提供与本公开有关的背景信息,而不必然构成现有技术。上世纪80年代LegerosR、BrownWE和ChowLC等人成功研制出可自固化的磷酸钙的骨水泥(CalciumPhosphateCement,CPC),开启了人们对于新型非陶瓷型CPC类人工骨修复材料的研究热潮。目前,研究人员对开发基于CPC的兴趣不断增加,是由于它们与人体骨质的矿物成分有许多共同的组成成分,并且其具有的生物降解性,生物活性和骨传导性是医学临床应用必不可少的要素。也是因为其可任意塑形并能够在体温和生理环境下自行固化,成为制备人工骨和填充被修复骨腔的重要材料,在临床上得到了广泛应用。据本公开发明人所知,目前对于骨水泥改性,以添加微量元素为例,利用Sr、Mg、Zn、Ag等元素对其进行改性。Sr元素改性的CPC骨水泥植入动物体内后,Sr离子极易从材料中析出,能够抑制破骨细胞的活动,促进新骨组织的形成,利于植入体周围骨组织的生长。Mg元素能够影响CPC骨水泥的固化速率、HA的结晶生长过程以及材料的机械强度,在骨水泥中添加合适的Mg元素有助于改善材料的机械性能和生物学性能。LiXia等人研究了Zn离子对CPC骨水泥的影响,兔子股骨植入试验结果表明含Zn的骨水泥能够促进成骨细胞的形成、生长,但是Zn元素的含量过高会导致手术部位的感染。感染是整形外科手术所面临的最大问题之一,它会加剧病人的痛苦并可能导致手术的失败。在CPC骨水泥中掺杂少量Ag离子对于手术后的感染有很好的预防作用,Ag离子从水泥中析出后能够增加材料的抗细菌能力。技术实现要素:为了拓展提升磷酸钙骨水泥的承重力学性能、生物活性、降低临床免疫排斥的可能性,本公开的目的是提供硅磷酸钙基体粉料及制备方法、骨修复材料及制备方法,该硅磷酸钙基体粉料能够在体温和生理环境下自行固化,并且形成的骨修复材料能够达到承重骨所需的要求。为了实现上述目的,本公开的技术方案为:第一方面,一种硅磷酸钙基体粉料,为α-硅磷酸三钙,其中,钙元素与磷元素的摩尔比为1.48~1.52:1,钙元素与硅元素的摩尔比为1:0.0326~0.0340。本公开经过实验表明,以钙元素与磷元素的摩尔比为1.48~1.52:1时,硅元素的添加量影响硅磷酸钙基体粉料形成骨水泥的力学性能,而当钙元素与硅元素的摩尔比为1:0.0326~0.0340,其制备的骨水泥的力学性能明显增强。第二方面,一种硅磷酸钙基体粉料的制备方法,以磷酸氢钙、碳酸钙和硅酸钙作为原料,混合均匀后,煅烧获得,磷酸氢钙、碳酸钙和硅酸钙的摩尔比为1:0.436~0.464:0.0485~0.0515。第三方面,一种骨修复材料(骨水泥)的制备方法,将上述硅磷酸钙基体粉料加入至固化液中进行固化成型,所述固化液为模拟体液(SBF)。经过实验表面,以模拟体液为固化液固化成型的骨修复材料具有显著的力学性能,其在0.64cm2的受力面积上可承受1800.9605N的压力。第四方面,一种上述骨修复材料的制备方法获得的骨修复材料。本公开的有益效果为:(1)本公开提供的硅磷酸钙基体粉料,硅元素的含量为3.26~3.40%时,可有效抑制无硅烧结伴生物羟基磷灰石(HA)的生成,也提升了α-TCP向β-TCP转变的温度,保证α-TCP的纯度。(2)本公开提供的骨修复材料中新生成的HA相主要来自α-TCP的转化水解,3.26~3.40%含量硅元素的掺杂减缓了转化HA的进程,使成品Si-CPC形成多相固溶体,从而使得骨修复材料的力学性能得到增强。(3)本公开3.26~3.40%含量的Si元素的引入,使骨水泥的孔隙率略有增加。2000nm左右长度的针状HA晶须得以生成,形成晶须增韧的效果。晶粒密度分别较为均匀,晶粒之间连接成片,相对于无硅磷酸钙骨水泥形貌更加致密,使得骨修复材料的力学性能得到进一步增强。(4)本公开以模拟体液固化硅磷酸钙骨修复材料,力学性能承重可达到0.64cm2的受力面积上可承受1800.9605N的压力,即35.829MPa。为实验对照组不含硅元素的磷酸钙骨修复材料承重16.89MPa力学性能的2.12倍。(5)模拟人体环境在SBF浸泡14天的过程中,3.26~3.40%硅磷酸钙骨修复材料,内部晶粒晶界逐渐模糊,晶须不断生成,密度逐渐增大,使其可持续保持较好的力学性能,XRD衍射分析结果显示,其物相完全转变为HA,这与人骨成分相一致,生物相容性良好。(6)SBF浸泡过程中,SBF固化的硅磷酸钙骨修复材料,表面晶须先增长,浸泡3天后,开始发生晶粒表面晶须降解现象,骨修复材料内部亦出现降解孔洞。晶粒表面及间隙被骨修复材料在SBF中吸收的P、O、Ca、H等成分生成的新的羟基磷灰石所覆盖及填充,晶粒间变的连接成片。表明该骨修复材料降解反应与生成反应之间的动态变化,最终稳定于生成反应大于降解反应的良好生物性能,这可以促进人骨细胞的替换生长,最终达到骨修复的目的。(7)本公开制备方法简单、力学机械性能高、生物活性佳、修复效率高、实用性强,易于推广。附图说明构成本公开的一部分的说明书附图用来提供对本公开的进一步理解,本公开的示意性实施例及其说明用于解释本公开,并不构成对本公开的不当限定。图1为本公开实施例1~7制备的硅磷酸钙基体粉料的XRD图谱;图2为本公开实施例8~14制备的骨水泥的XRD图谱;图3为本公开实施例11制备的SBF-4A经14天模拟人体环境浸泡实验的XRD图谱;图4为本公开实施例8制备的SBF-1A的截断中心部位的扫描电镜照片,(a)为500倍,(b)为1000倍,(c)为2000倍,(d)为5000倍;图5为本公开实施例11制备的SBF-4A的截断中心部位的扫描电镜照片,(a)为1000倍,(b)为2000倍,(c)为5000倍,(d)为8000倍;图6为本公开实施例11制备的SBF-4A的元素面扫描图,Ca、P、O三种元素代表着羟基磷灰石及磷酸三钙的分布,Si元素代表着CaSiO3的分布,Cl元素表示模拟体液的分布;图7为本公开实施例11制备的SBF-4A的经14天模拟人体环境浸泡实验过程的2000倍的扫描电镜照片,(a)为浸泡1天的截断面中心的微观形,(b)为浸泡1天的侧表面的微观形貌,(c)为浸泡3天的截断面中心的微观形,(d)为浸泡3天的侧表面的微观形貌,(e)为浸泡7天的截断面中心的微观形,(f)为浸泡7天的侧表面的微观形貌,(g)为浸泡14天的截断面中心的微观形,(h)为浸泡14天的侧表面的微观形貌;图8为本公开实施例11制备的SBF-4A的经14天模拟人体环境浸泡实验过程的5000倍的扫描电镜照片,(a)为浸泡1天的截断面中心的微观形,(b)为浸泡1天的侧表面的微观形貌,(c)为浸泡3天的截断面中心的微观形,(d)为浸泡3天的侧表面的微观形貌,(e)为浸泡7天的截断面中心的微观形,(f)为浸泡7天的侧表面的微观形貌,(g)为浸泡14天的截断面中心的微观形,(h)为浸泡14天的侧表面的微观形貌;图9为本公开实施例8~14制备的骨水泥的力学性能柱状图;图10为本公开实施例8~14制备的骨水泥的孔隙率的趋势图。具体实施方式应该指出,以下详细说明都是示例性的,旨在对本公开提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本公开所属
技术领域:
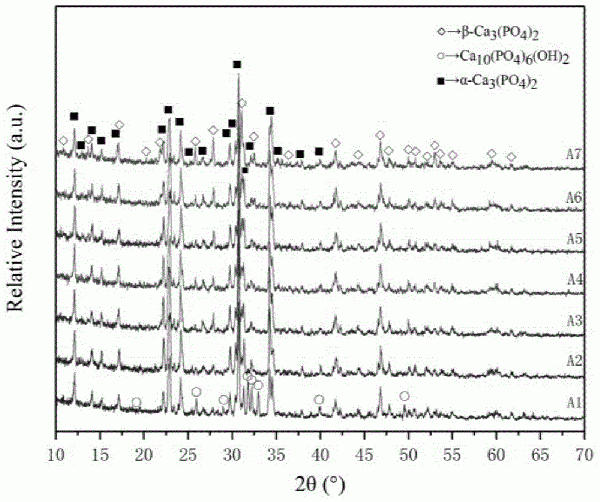
的普通技术人员通常理解的相同含义。需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本公开的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。本公开中硅含量是指硅元素摩尔量与钙元素摩尔量的商。为了拓展提升磷酸钙骨水泥的承重力学性能、生物活性、降低临床免疫排斥的可能性,本公开提出了硅磷酸钙基体粉料及制备方法、骨修复材料及制备方法。本公开的一种典型实施方式,提供了一种硅磷酸钙基体粉料,为α-硅磷酸三钙,其中,钙元素与磷元素的摩尔比为1.48~1.52:1,钙元素与硅元素的摩尔比为1:0.0326~0.0340。本公开经过实验表明,以钙元素与磷元素的摩尔比为1.48~1.52:1时,硅元素的添加量影响硅磷酸钙基体粉料形成骨水泥的力学性能,而当钙元素与硅元素的摩尔比为1:0.0326~0.0340,其制备的骨水泥的力学性能明显增强。该实施方式的一种或多种实施例中,所述硅磷酸钙基体粉料的粒径为70~80μm。本公开的另一种实施方式,提供了一种硅磷酸钙基体粉料的制备方法,以磷酸氢钙、碳酸钙和硅酸钙作为原料,混合均匀后,煅烧获得,磷酸氢钙、碳酸钙和硅酸钙的摩尔比为1:0.436~0.464:0.0485~0.0515。该实施方式的一种或多种实施例中,混合方式为球磨。该系列实施例中,球磨过程中添加乙醇。保证原料混合的均匀性。该系列实施例中,球磨的速度为200~300r/min,球磨时间为1~2h。该实施方式的一种或多种实施例中,煅烧的温度为1240~1256℃,煅烧时间为2~3h。该实施方式的一种或多种实施例中,煅烧后进行研磨。本公开的第三种实施方式,提供了一种骨修复材料的制备方法,将上述硅磷酸钙基体粉料加入至固化液中进行固化成型,所述固化液为模拟体液。经过实验表面,以模拟体液为固化液固化成型的骨修复材料具有显著的力学性能,其在0.64cm2的受力面积上可承受1800.9605N的压力。该实施方式的一种或多种实施例中,固化液与硅磷酸钙基体粉料的液固比为0.4~1.0:1,mL:g。该系列实施例中,固化液与硅磷酸钙基体粉料的液固比为0.4~0.5:1,mL:g。该实施方式的一种或多种实施例中,所述固化成型的过程为:将硅磷酸钙基体粉料和固化液加入至模具中,脱模后放置在人体温度及100%气体湿度下处理,然后烘干。该系列实施例中,在模具中成型时间为15~20min。该系列实施例中,脱模后的处理时间为12~72h。本公开的第四种实施方式,提供了一种上述骨修复材料的制备方法获得的骨修复材料。为了使得本领域技术人员能够更加清楚地了解本公开的技术方案,以下将结合具体的实施例详细说明本公开的技术方案。实施例1硅磷酸钙基体粉料A1的制备。将二水磷酸氢钙(CaHPO4·2H2O、DCPD)与碳酸钙(CaCO3、CC)按2:1的比例,分批次装入球磨罐。加入无水乙醇用于保证浆料的混合均匀性,在球磨机中以速度225r/min球磨1小时,进而移入烘干箱,65℃下干燥12小时。将粉料移进100mL刚玉坩埚后,在箱式气氛炉中以4℃/min的升温速度,加热至1250℃后烧结2.5小时,随即在常温空气下进行急冷。经玛瑙研钵研磨后,过200目标准检验筛,获得硅磷酸钙基体粉料,记为A1,A1粉料颗粒尺寸约74μm,移入50mL离心管密封保存。实施例2硅磷酸钙基体粉料A2的制备。将二水磷酸氢钙(CaHPO4·2H2O、DCPD)与碳酸钙(CaCO3、CC)及硅酸钙(CaSiO3、CS)按2:0.96:0.04的比例,分批次装入球磨罐。加入适量的无水乙醇用于保证浆料的混合均匀性,在球磨机中以速度225r/min球磨1小时,进而移入烘干箱,65℃下干燥12小时。将粉料移进100mL刚玉坩埚后,在箱式气氛炉中以4℃/min的升温速度,加热至1250℃后烧结2.5小时,随即在常温空气下进行急冷。经玛瑙研钵研磨后,过200目标准检验筛,获得硅磷酸钙基体粉料(Si占比1.33%),记为A2,A2粉料颗粒尺寸约74μm,移入50mL离心管密封保存。实施例3硅磷酸钙基体粉料A3的制备。将二水磷酸氢钙(CaHPO4·2H2O、DCPD)与碳酸钙(CaCO3、CC)及硅酸钙(CaSiO3、CS)按2:0.92:0.08的比例,分批次装入球磨罐。加入适量的无水乙醇用于保证浆料的混合均匀性,在球磨机中以速度225r/min球磨1小时,进而移入烘干箱,65℃下干燥12小时。将粉料移进100mL刚玉坩埚后,在箱式气氛炉中以4℃/min的升温速度,加热至1250℃后烧结2.5小时,随即在常温空气下进行急冷。经玛瑙研钵研磨后,过200目标准检验筛,获得硅磷酸钙基体粉料(Si占比2.67%),记为A3,A3粉料颗粒尺寸约74μm,移入50mL离心管密封保存。实施例4硅磷酸钙基体粉料A4的制备。将二水磷酸氢钙(CaHPO4·2H2O、DCPD)与碳酸钙(CaCO3、CC)及硅酸钙(CaSiO3、CS)按2:0.90:0.10的比例,分批次装入球磨罐。加入适量的无水乙醇用于保证浆料的混合均匀性,在球磨机中以速度225r/min球磨1小时,进而移入烘干箱,65℃下干燥12小时。将粉料移进100mL刚玉坩埚后,在箱式气氛炉中以4℃/min的升温速度,加热至1250℃后烧结2.5小时,随即在常温空气下进行急冷。经玛瑙研钵研磨后,过200目标准检验筛,获得硅磷酸钙基体粉料(Si占比3.33%),记为A4,A4粉料颗粒尺寸约74μm,移入50mL离心管密封保存。实施例5硅磷酸钙基体粉料A5的制备。将二水磷酸氢钙(CaHPO4·2H2O、DCPD)与碳酸钙(CaCO3、CC)及硅酸钙(CaSiO3、CS)按2:0.88:0.12的比例,分批次装入球磨罐。加入适量的无水乙醇用于保证浆料的混合均匀性,在球磨机中以速度225r/min球磨1小时,进而移入烘干箱,65℃下干燥12小时。将粉料移进100mL刚玉坩埚后,在箱式气氛炉中以4℃/min的升温速度,加热至1250℃后烧结2.5小时,随即在常温空气下进行急冷。经玛瑙研钵研磨后,过200目标准检验筛,获得硅磷酸钙基体粉料(Si占比4.00%),记为A5,A5粉料颗粒尺寸约74μm,移入50mL离心管密封保存。实施例6硅磷酸钙基体粉料A6的制备。将二水磷酸氢钙(CaHPO4·2H2O、DCPD)与碳酸钙(CaCO3、CC)及硅酸钙(CaSiO3、CS)按2:0.84:0.16的比例,分批次装入球磨罐。加入适量的无水乙醇用于保证浆料的混合均匀性,在球磨机中以速度225r/min球磨1小时,进而移入烘干箱,65℃下干燥12小时。将粉料移进100mL刚玉坩埚后,在箱式气氛炉中以4℃/min的升温速度,加热至1250℃后烧结2.5小时,随即在常温空气下进行急冷。经玛瑙研钵研磨后,过200目标准检验筛,获得硅磷酸钙基体粉料(Si占比5.33%),记为A6,A6粉料颗粒尺寸约74μm,移入50mL离心管密封保存。实施例7硅磷酸钙基体粉料A7的制备。将二水磷酸氢钙(CaHPO4·2H2O、DCPD)与碳酸钙(CaCO3、CC)及硅酸钙(CaSiO3、CS)按2:0.80:0.20的比例,分批次装入球磨罐。加入适量的无水乙醇用于保证浆料的混合均匀性,在球磨机中以速度225r/min球磨1小时,进而移入烘干箱,65℃下干燥12小时。将粉料移进100mL刚玉坩埚后,在箱式气氛炉中以4℃/min的升温速度,加热至1250℃后烧结2.5小时,随即在常温空气下进行急冷。经玛瑙研钵研磨后,过200目标准检验筛,获得硅磷酸钙基体粉料(Si占比6.67%),记为A7,A7粉料颗粒尺寸约74μm,移入50mL离心管密封保存。模拟体液的制备过程:向1000mL的烧杯中添加750mL去离子水,定温36.5℃,将7.996g氯化钠、0.350g碳酸氢钠、0.224g氯化钾、0.228g三水合磷酸氢二钾、40mL1mol/L盐酸、0.305g六水合氯化镁、0.278g氯化钙和0.071g硫酸钠加入至烧杯中溶解,然后,每次放入少于1gTris(CH2OH)3CXH2,溶解,共加入6.051gTris(CH2OH)3CXH2,然后定容至1000mL,最后采用1mol/L盐酸调节pH至7.25。实施例8骨水泥SBF-1A的制备。将实施例1的A1粉料与模拟体液以固液比为1:0.45(g/mL)的比例添加至坩埚中混合均匀后,立即注入直径为8mm、高度为12mm的圆柱孔304不锈钢模具,人工使用T型冲针稍施压力压制成型。同时制备6个骨水泥柱,均需在15-20分钟左右脱模,随即移入培养皿,置于37℃恒温水浴锅上层,保持100%气体湿度48小时后,在烘干箱中烘干,获得骨水泥,记为SBF-1A,妥存,以备测试。实施例9骨水泥SBF-2A的制备。将实施例2的A2粉料与模拟体液以固液比为1:0.45(g/mL)的比例添加至坩埚中混合均匀后,立即注入直径为8mm、高度为12mm的圆柱孔304不锈钢模具,人工使用T型冲针稍施压力压制成型。同时制备6个骨水泥柱,均需在15-20分钟左右脱模,随即移入培养皿,置于37℃恒温水浴锅上层,保持100%气体湿度48小时后,在烘干箱中烘干,获得骨水泥,记为SBF-2A,妥存,以备测试。实施例10骨水泥SBF-3A的制备。将实施例3的A3粉料与模拟体液以固液比为1:0.45(g/mL)的比例添加至坩埚中混合均匀后,立即注入直径为8mm、高度为12mm的圆柱孔304不锈钢模具,人工使用T型冲针稍施压力压制成型。同时制备6个骨水泥柱,均需在15-20分钟左右脱模,随即移入培养皿,置于37℃恒温水浴锅上层,保持100%气体湿度48小时后,在烘干箱中烘干,获得骨水泥,记为SBF-3A,妥存,以备测试。实施例11骨水泥SBF-4A的制备。将实施例4的A4粉料与模拟体液以固液比为1:0.45(g/mL)的比例添加至坩埚中混合均匀后,立即注入直径为8mm、高度为12mm的圆柱孔304不锈钢模具,人工使用T型冲针稍施压力压制成型。同时制备6个骨水泥柱,均需在15-20分钟左右脱模,随即移入培养皿,置于37℃恒温水浴锅上层,保持100%气体湿度48小时后,在烘干箱中烘干,获得骨水泥,记为SBF-4A,妥存,以备测试。实施例12骨水泥SBF-5A的制备。将实施例5的A5粉料与模拟体液以固液比为1:0.45(g/mL)的比例添加至坩埚中混合均匀后,立即注入直径为8mm、高度为12mm的圆柱孔304不锈钢模具,人工使用T型冲针稍施压力压制成型。同时制备6个骨水泥柱,均需在15-20分钟左右脱模,随即移入培养皿,置于37℃恒温水浴锅上层,保持100%气体湿度48小时后,在烘干箱中烘干,获得骨水泥,记为SBF-5A,妥存,以备测试。实施例13骨水泥SBF-6A的制备。将实施例6的A6粉料与模拟体液以固液比为1:0.45(g/mL)的比例添加至坩埚中混合均匀后,立即注入直径为8mm、高度为12mm的圆柱孔304不锈钢模具,人工使用T型冲针稍施压力压制成型。同时制备6个骨水泥柱,均需在15-20分钟左右脱模,随即移入培养皿,置于37℃恒温水浴锅上层,保持100%气体湿度48小时后,在烘干箱中烘干,获得骨水泥,记为SBF-6A,妥存,以备测试。实施例14骨水泥SBF-7A的制备。将实施例7的A7粉料与模拟体液以固液比为1:0.45(g/mL)的比例添加至坩埚中混合均匀后,立即注入直径为8mm、高度为12mm的圆柱孔304不锈钢模具,人工使用T型冲针稍施压力压制成型。同时制备6个骨水泥柱,均需在15-20分钟左右脱模,随即移入培养皿,置于37℃恒温水浴锅上层,保持100%气体湿度48小时后,在烘干箱中烘干,获得骨水泥,记为SBF-7A,妥存,以备测试。Si占比的计算公式为:性能测试方法:X射线衍射分析:采用日本日本津岛公司(SHIMADZU)生产的XRD-6100型X射线衍射仪分析样品物相组成,管电压40kV,管电流40mA,扫描范围10°~90°,扫描速度4°/min。将试样磨成粉后置于测试模具内压平后,进行测试分析。扫描电子显微镜与成分分析:采用日本日立公司(HITACHI)生产的S-3400N型扫描电镜(Scanningelectronmicroscope,SEM)观察样品表面微观组织形貌,扫描电镜附件能谱仪(Energydispersivespectrometer,EDS)附件由HORIBA公司生产,型号为EMAXX-act,主要用于样品表面成分分析。由于样品不导电,因此,测试之前需要对其进行喷金处理,喷金时间2分钟,喷金设备为北京中科科仪技术发展有限责任公司研制的KYKYSBC-12型离子溅射仪。抗压强度的测定:采用美特斯工业系统(中国)有限公司CMT5105型号的微机控制电子试验机进行骨水泥试样抗压强度测试,加载速度为0.5mm/min。定力衰减起始判断力为30N。抗压强度计算公式为:σ=4P/πD2式中,D—试样直径8mm;σ—压缩强度,mPa;P—测试最大压力,N。由于样品上下表面水平度有误差,同一配方样品测定2个以上平行样品,求取平均值。孔隙率测试:孔隙率P的测定是基于阿基米德原理,试验中采用浸水法测定孔隙率。首先将200mL烧杯中,注入去离子水,接着将烧杯加热至沸腾,然后将单个样品置于烧杯,保持100摄氏度水温1h,使去离子水完全浸透样品。停止加热至室温后,用镊子取出样品,用吸水纸吸去样品表面的水,快速称量样品的浸水水泥质量记为m1;通过天平附件吊篮,使试样悬浮水中,称取含水样品在水中的悬浮质量记为m2;最后将样品干燥称重,记为m3。计算孔隙率公式如下:结果分析:如图1所示,A1对比单斜晶系的α-磷酸三钙(α-Ca3(PO4)2、α-TCP)(PDF#70-0364、PDF#29-0359、PDF#09-0348)可知,磷酸氢钙(CaHPO4·2H2O、DCPD)与碳酸钙(CaCO3、CC)在1250℃下保温烧结2.5小时后随即空冷可得到几乎不含β相的α-TCP,在室温下稳定性较好。伴随生成部分羟基磷灰石(Ca10(PO4)6(OH)2、HA)(PDF#73-1731、PDF#72-1243、PDF#09-0432)该相为后期制作骨水泥生成的主要成分,即有益相。同时上述α-TCP的PDF卡片主要识别区域在2θ角为27.5度-37.5度之间,根据生成物及利用JADE软件分析,2θ为41.7°处可能含有α-TCP的峰形。A2-A7同样根据XRD原始数据在JADE中的分析可知,微小的硅含量即可保证生成物中不含有HA相,在2θ为30.7°处的α-TCP主峰(PDF#29-0359中为034晶面),其强度自A1至A4时平缓上升,随后A5-A7相对明显下降,A7与A4的强度比约为0.78,说明α-TCP的含量先增加后稍降低;对比菱方晶系的β-磷酸三钙(β-Ca3(PO4)2、β-TCP)(PDF#09-0169、PDF#86-1585),在2θ为31.2°处β-TCP峰(PDF#09-0169中为0210晶面),其相对强度自A3-A7开始出现并逐渐显著增强,即表明β-TCP的生成及其含量逐渐增加。综上可知,掺硅元素进入α-TCP,可有效抑制羟基磷灰石的生成,但是随着掺入硅元素的增多,提升了α-TCP向β-TCP转变的温度,即α-TCP的生成纯度有所提高。如图2所示,SBF-1A的X射线衍射图谱曲线表明,α-TCP几乎完全转化为了(CD)HA,其峰型尖锐且清晰,表明(CD)HA结晶度良好。SBF-2A与SBF-1A对比明显看出α-TCP的衍射峰强度最高,表明微含量的Si元素引起α-TCP向(CD)HA转换不完全。随着Si元素的继续引入,α-TCP衍射峰强度曲折中降低,SBF-4A的α-TCP衍射峰强度,相对于SBF-3A、SBF-5A中的α-TCP衍射峰有所升高,SBF-3A、SBF-5A的α-TCP衍射峰强度大致相同。这表明SBF-4A因为特定Si元素含量的掺杂,其晶体结构发生不随着Si元素含量变化规律而变动,而出现α-TCP衍射峰降低的特殊情况。SBF-4A相中(CD)HA的衍射峰型尖锐,底部钝化宽化,结合SEM分析,这是由于纳米晶须的存在。直至SBF-7A时,α-TCP几乎消失,而(CD)HA峰底最为宽泛,结合SEM分析这是由于晶体表面成层片状结构。其中可能包含化合物(Ca2SiO4)0.05Ca3(PO4)2(PDF#49-1674),Ca5(PO4)2SiO4(PDF#73-1181)的衍射峰,硅元素未能对α-TCP水化生成(CD)HA产生阻碍的原因是生成了含硅化合物。同时,β-TCP的峰型及相对衍射强度仍与图1中的趋势相似,其在CPC生成过程中几乎未发生转化。在以模拟体液为固化液时,HA的转化量明显增加,模拟体液中的Ca元素补充到了α-TCP的水化过程中,即(CD)HA含量相对减少。因而,从图1~图2可以得出,在模拟体液中,引入硅元素含量较低时,部分阻碍α-TCP向HA的水解转化。随着Si元素的增加,向着HA转化率明显增加,模拟体液补充生成HA所需的Ca元素。常温下水化,硅元素存在时,β-TCP几乎不能水解。如图3所示,代号为1Day至14Day的各物相三强峰的主要识别区域在2θ角为27.5度-37.5度之间,采取最强峰及次强峰为该物相的主要指示峰。α-TCP主要指示峰为(PDF#09-0348)正交晶系2θ为30.73度处的(170)晶面及2θ为34.16度的(043)晶面,经Jade软件测算其晶粒度大小为>1000nm。β-TCP主要指示峰为(PDF#09-0169)三方晶系2θ为31.07度的(0210)晶面及2θ为34.41度的(220)晶面经Jade软件测算其晶粒度大小>1000nm。CDHA与HA的位置、晶系、晶面几乎相同,HA主要指示峰为(PDF#72-1243)六方晶系2θ为31.84度的(211)晶面及2θ为32.97度的(300)晶面,经Jade软件测算其晶粒度大小约为100nm。与原始SBF固化后的硅磷酸钙骨水泥成品物相成分相比,在模拟体液当中,浸泡1天时,α-TCP即几乎完全转化生成为羟基磷灰石(HA),浸泡3天时,物相分析显示α-TCP相复现;浸泡7天及14天,羟基磷灰石相衍射峰强度逐渐升高,峰型愈发尖锐,衍射峰面积增加,表明羟基磷灰石晶体含量大量增加且晶体成型度良好,其晶粒尺寸变小。与此同时,可以观察到α-TCP衍射峰强度最终不可见,同样说明α-TCP已经完全转化为HA相。在实际浸泡过程中,每两天更换一次新的模拟体液,以模拟人体内体液不断新陈代谢,同时每两天测试模拟体液pH值变化情况,如表1所示。发现1至14天的浸泡溶液pH值逐渐减少,7天之后趋于稳定值,这说明SBF浸泡下的硅磷酸钙骨水泥化学反应趋势趋于稳定。表1浸泡过程中模拟体液的pH值前4天更换模拟体液时,可以观察到,溶液一致清澈,无可见颜色变化。根据其pH值得变化,可以解释浸泡前3天,模拟体液不断浸入,发生了一个物相还原现象,即骨水泥中α-TCP的复现,这可能是由于该pH下,更适合羟基磷灰石向α-TCP的逆向生成。将SBF浸泡后的骨水泥成品及时取出干燥,与其原始质量对比,其质量变化如表2所示。表2SBF浸泡后的骨水泥成品与其原始质量的对比浸泡时间1Day3Day7Day14Day质量增长比1.86%2.82%2.57%4.01%表明随着浸泡时间的推移,SBF中的物质被硅磷酸钙骨水泥不断吸收,质量不断增加。这与XRD衍射图谱所指示的物相反应所应具备的质量变化条件一致。综上,可以得到结论,经SBF固化的硅磷酸钙骨水泥,在模拟体液当中,随着时间的推移,α-TCP转化为HA相的趋势先减小后逐渐加深,并最终完全转化,这与人骨主要成分相符。pH值逐渐降低并趋于稳定,总体呈中性偏弱碱,可以很好的与人体液环境兼容。骨水泥质量不断增加,表明其可生长性良好。如图4所示,500倍至1000倍当中,其晶粒外观较为规则成团状或柱状,晶粒大小在5-10μm之间,其密度分别较为均匀,空隙孔径大小相近,表明经A1粉体固化生成过程中,其晶体结构有序转变,晶体成型度良好。SBF-4A是以含硅元素含量为3.3%的磷酸三钙粉末经SBF固化生成的硅磷酸钙骨水泥,在其XRD衍射图谱显示其主要物质为羟基磷灰石及尚未转化完成的α-TCP。如图5所示,从1000倍至2000倍形貌图当中,可以看出,其晶粒间相连部位的区分度不似SBF-1A中明显,截断过程中,其截断面稍显不平整,表明其内部抵抗应变力的能力不均匀。晶粒密度分别较为均匀,晶粒之间连接成片,部分空隙消失,相对于SBF-1A形貌更加致密。在5000倍及8000倍下,可以观察到晶体直径约5~10μm,晶体之间结合部位交错重叠了大量的羟基磷灰石晶须,晶须长度在2000nm左右,与SBF-1A比较后,即可得到Si元素的掺入,是晶须生成的原因。晶须的生成,便是各晶粒间区分度不明显的原因,极大地增强了硅磷酸钙骨水泥的力学性能。如图6所示,Ca、P、O、Si、Cl均可以均匀分布于整个面内,这表示在烧结前湿混粉末的均匀性良好,烧结过程中化学反应充分,制成骨水泥过程中,粉末与SBF固化液搅拌均匀。SBF固化的硅磷酸钙骨水泥中使用的SBF固化液,是最接近人体液成分的配比的无机溶液,与人体发生排斥反应的可能性最小。在此基础上开发有较强的力学性能的骨水泥相对可医用性最好。SBF固化的A4原始粉末骨水泥,初始时,其力学性能为在0.64cm2的承重面积下可承受约180.1kg的重量。在浸泡过程中,如图7~8所示,整体外观未发生变形,骨水泥的力学性能保持良好。由图7(a)、图7(c)、图7(e)、图7(g)可以看出,在2000倍下,晶粒之间的晶界越发模糊、堆积密度逐渐加大,晶须长度逐渐加大。如图8(a)、图8(c)、图8(e)、图8(g)所示,放大至5000倍下,可以观察到其晶粒尺寸从浸泡1天至浸泡14天的明显变化,由于晶须密度及长度的的增大,晶粒间的区分困难。晶须密度逐渐上升的同时,晶须尺寸从浸泡1天的约为1000nm开始生长至14天的2000nm左右,这说明晶体内部由于SBF的浸泡,促进了羟基磷灰石晶须生成与生长。同时晶粒间的空隙孔径逐渐缩小,至14天时,基本被晶须所覆盖。值得突出说明的是,骨水泥内部从第3天开始出现了被降解的孔洞,至14天时出现明显被降解的孔洞。如图7(b)、图7(d)、图7(f)、图7(h)所示,在2000倍下,随着SBF浸泡过程的进行,表面形貌发生了巨大变化。第1天的晶粒表面的晶须长度呈爆发式增长,到3天时晶须完全被降解,表面部分覆盖了新的羟基磷灰石层。7天、14天时,晶粒间隙逐渐被填充,晶粒表面的棱角逐渐被溶解,变得圆润,羟基磷灰石涂层越来越厚,表面晶粒已连接成片。如图8(b)、图8(d)、图8(f)、图8(h)所示,放大至5000倍后观察,浸泡1天时表面产生的羟基磷灰石晶须约为7000nm左右,晶须直径约为500-1000nm,显得十分坚挺。在第3天时,由于骨水泥已经被完全浸透,模拟体液与骨水泥发生离子交换,pH值降低,羟基磷灰石的生成反应与降解反应同时在发生,骨水泥内部旧的羟基磷灰石在外部环境改变的情况下,部分转变为磷酸三钙。外表面的羟基磷灰石晶须被溶解。在随后的浸泡过程中,晶粒之间发生融合,初始晶界消失。新的羟基磷灰石生成速率大于降解速率,至7天时,骨水泥的XRD衍射分析显示其完全转变为了羟基磷灰石。至14天时,融合后的晶粒尺寸约为10000nm-20000nm。晶粒间的空隙孔洞亦是在14天的浸泡过程中,随着晶粒的长大逐渐消失。与此同时,晶粒表面出现的扁圆状孔洞逐渐加深,这说明羟基磷灰石发生降解现象一直在进行,降解后的离子溶解到SBF当中。这种生成与降解同时存在的状态,为生物自身生成骨细胞进行替代人工骨水泥提供了有利条件。至此,可以得出结论,SBF浸泡过程中,SBF固化的硅磷酸钙骨水泥,内部晶粒晶界逐渐模糊,晶须不断生成,密度逐渐增大,使其可持续保持较好的力学性能,促进其更好的满足人工骨水泥在骨修复、骨承重的力学性能要求。SBF浸泡过程中,SBF固化的硅磷酸钙骨水泥,表面晶须先增长,浸泡3天后,开始发生晶粒表面晶须降解现象,水泥内部亦出现降解孔洞。晶粒表面及间隙被骨水泥在SBF中吸收的P、O、Ca、H等成分生成的新的羟基磷灰石所覆盖及填充,晶粒间变的连接成片。表明该人工骨水泥降解反应与生成反应动态变化,最终稳定于生成反应大于降解反应的良好生物性能,促进人骨细胞的替换生长,最终达到骨修复的目的。从图9可以清晰的看出,当硅含量为3.33%的硅磷酸钙骨水泥,其力学性能明显升高,结合XRD/SEM分析,表明Si元素的掺入量为最优状态。力学性能为不含硅元素的磷酸钙骨水泥的2.12倍。由图10可知,随着硅含量的增加,孔隙率成先增加后减少的趋势,整体变数值化并不明显。对比SBF-1A及SBF-4A,此时孔隙率增长约1%,与力学性能相比增加112%,没有明显关联。结论:(1)模拟体液自固化硅磷酸钙骨修复材料,力学性能承重可达到0.64cm2的受力面积上可承受1800.9605N的压力,即35.829MPa。为实验对照组不含硅元素的磷酸钙骨水泥承重16.89MPa力学性能的2.12倍。(2)掺3.33%含量硅元素烧结而成的α-TCP,可有效抑制无硅烧结伴生物羟基磷灰石(HA)的生成,也提升了α-TCP向β-TCP转变的温度,保证α-TCP的纯度。(3)固化骨水泥中新生成的HA相主要来自α-TCP的转化水解,3.33%含量硅元素的掺杂减缓了转化HA的进程,使成品Si-CPC形成多相固溶体,这是力学性能得以加强的原因之一。(4)3.33%含量的Si元素的引入,使骨水泥的孔隙率略有增加。2000nm左右长度的针状HA晶须得以生成,形成晶须增韧的效果。晶粒密度分别较为均匀,晶粒之间连接成片,相对于无硅磷酸钙骨水泥形貌更加致密,这亦是力学性能得以加强的原因。(5)模拟人体环境在SBF浸泡14天的过程中,3.33%硅磷酸钙骨水泥,内部晶粒晶界逐渐模糊,晶须不断生成,密度逐渐增大,使其可持续保持较好的力学性能,XRD衍射分析结果显示,其物相完全转变为HA,这与人骨成分相一致,生物相容性良好!(6)SBF浸泡过程中,SBF固化的硅磷酸钙骨水泥,表面晶须先增长,浸泡3天后,开始发生晶粒表面晶须降解现象,水泥内部亦出现降解孔洞。晶粒表面及间隙被骨水泥在SBF中吸收的P、O、Ca、H等成分生成的新的羟基磷灰石所覆盖及填充,晶粒间变的连接成片。表明该人工骨水泥降解反应与生成反应之间的动态变化,最终稳定于生成反应大于降解反应的良好生物性能,这可以促进人骨细胞的替换生长,最终达到骨修复的目的。以上所述仅为本公开的优选实施例而已,并不用于限制本公开,对于本领域的技术人员来说,本公开可以有各种更改和变化。凡在本公开的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本公开的保护范围之内。当前第1页1 2 3 
技术特征:
1.一种硅磷酸钙基体粉料,其特征是,为α-硅磷酸三钙,其中,钙元素与磷元素的摩尔比为1.48~1.52:1,钙元素与硅元素的摩尔比为1:0.0326~0.0340。
2.如权利要求1所述的硅磷酸钙基体粉料,其特征是,所述硅磷酸钙基体粉料的粒径为70~80μm。
3.一种硅磷酸钙基体粉料的制备方法,其特征是,以磷酸氢钙、碳酸钙和硅酸钙作为原料,混合均匀后,煅烧获得,磷酸氢钙、碳酸钙和硅酸钙的摩尔比为1:0.436~0.464:0.0485~0.0515。
4.如权利要求3所述的硅磷酸钙基体粉料的制备方法,其特征是,混合方式为球磨。
5.如权利要求4所述的硅磷酸钙基体粉料的制备方法,其特征是,球磨过程中添加乙醇;
或,球磨的速度为200~300r/min,球磨时间为1~2h。
6.如权利要求3所述的硅磷酸钙基体粉料的制备方法,其特征是,煅烧的温度为1240~1256℃,煅烧时间为2~3h。
7.一种骨修复材料的制备方法,其特征是,将权利要求1或2所述的硅磷酸钙基体粉料或权利要求3~6任一所述硅磷酸钙基体粉料的制备方法获得的硅磷酸钙基体粉料加入至固化液中进行固化成型,所述固化液为模拟体液。
8.如权利要求7所述的骨修复材料的制备方法,其特征是,固化液与硅磷酸钙基体粉料的液固比为0.4~1.0:1,mL:g;
优选的,固化液与硅磷酸钙基体粉料的液固比为0.4~0.5:1,mL:g。
9.如权利要求7所述的骨修复材料的制备方法,其特征是,所述固化成型的过程为:将硅磷酸钙基体粉料和固化液加入至模具中,脱模后放置在人体温度及100%气体湿度下处理,然后烘干;
优选的,在模具中成型时间为15~20min;
优选的,脱模后的处理时间为12~72h。
10.一种权利要求7~9任一所述的骨修复材料的制备方法获得的骨修复材料。
技术总结
本公开提供了硅磷酸钙基体粉料及制备方法、骨修复材料及制备方法,硅磷酸钙基体粉料,为α?硅磷酸三钙,其中,钙元素与磷元素的摩尔比为1.48~1.52:1,钙元素与硅元素的摩尔比为1:0.0326~0.0340。骨修复材料的制备过程为:将上述硅磷酸钙基体粉料加入至固化液中进行固化成型,所述固化液为模拟体液。本公开提供的硅磷酸钙基体粉料能够在体温和生理环境下自行固化,并且形成的骨修复材料能够达到承重骨所需的要求。
技术研发人员:王佃刚;明星辰;陈传忠;肖飞虹
受保护的技术使用者:山东大学
技术研发日:2019.07.12
技术公布日:2019.09.20
声明:
“硅磷酸钙基体粉料及制备方法、骨修复材料及制备方法与流程” 该技术专利(论文)所有权利归属于技术(论文)所有人。仅供学习研究,如用于商业用途,请联系该技术所有人。
我是此专利(论文)的发明人(作者)
 863
编辑:北方有色网
来源:山东大学
863
编辑:北方有色网
来源:山东大学



 咨询细节
咨询细节









































































 2026年08月06日 ~ 08日
2026年08月06日 ~ 08日 















